
SMART 2
A Comprehensive Analysis Tool for Bisulfite Sequencing Data


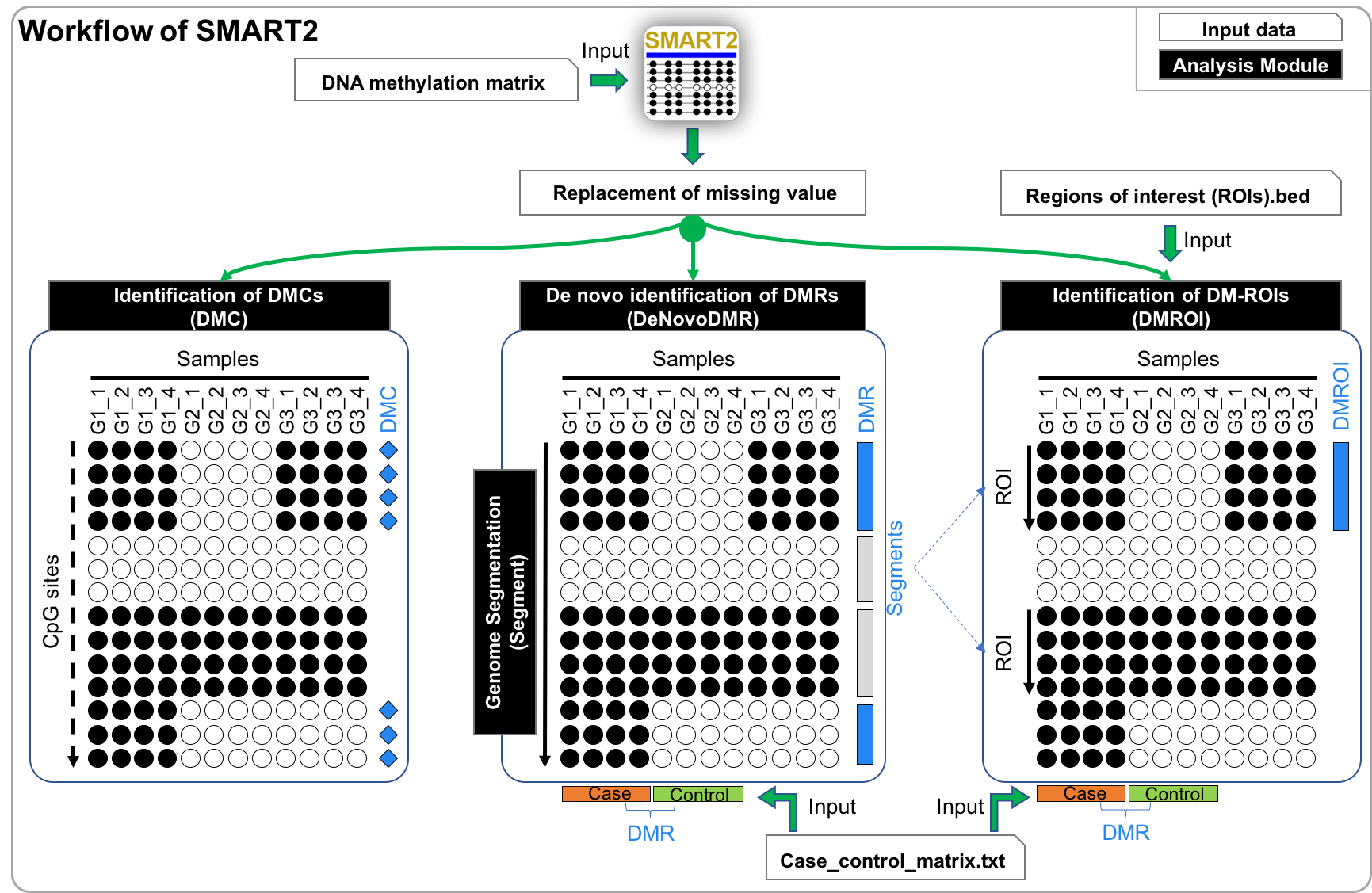
SMART2 is a newly developed tool for deep analysis of DNA methylation data detected by bisulfite sequencing platforms. This tool is focused on three main functions including de novo identification of differentially methylated regions (DMRs), genome segmentation, identification of DMRs from predefined regions of interest, and identification of differentially methylated CpG sites. In particular, SMART2 supports Case-Control DMR calling which is useful for disease methylome analsyis. It is known that DNA methylation plays important roles in the regulation of cell development and differentiation. DNA methylation/unmethylation mechanisms are common in all tissue/cell. However, different cell types with the same genome have different methylomes. Recently, high-throughput sequencing combining bisulfite treatment (Bisulfite-Seq) have been used to generate DNA methylomes from a wide range of human tissue/cell types at a genome-wide perspective. In order to de novo identify DMRs across different biological groups, entropy-based procedures facilitated the quantification of methylation specificity for each CpG and the determination of the Euclidean distance and similar entropy for each pair of neighboring CpGs. Subsequently, genome segmentation based on these quantified parameters segments the genome into primary segments comprising CpG sites with high methylation similarities across all groups. Further, the primary segments in close proximity and sharing similar methylation patterns were merged into larger segments of different types, including DMRs and non-DMRs which are identified based on methylation specificity and one-way ANOVA analysis. Eventually, the DMRs with specific hypo-/hypermethylation in the minority of groups, group-specific hypomethylation marks (HypoMarks) and the group-specific hypermethylation marks (HyperMarks), are identified using a statistical method. To facilitate the mining of methylation marks across cell types and species. In addition, SMART2 also supports the identification of DMRs from pre-defined regions of interest and differentially methylated CpG sites.
The package of SMART2 (Version 2.2.8) is available in the Python Package Index (https://pypi.org/project/SMART-BS-Seq/).
You are welcome to submit any issues at the (Github) or (Google group).
The source code of SMART is stored on PyPI - the Python Package Index which is a repository of software for the Python programming language. To use SMART package from this index either "pip install SMART-BS-Seq" (get pip) or download, unpack and "python setup.py install" it.
SMART is developed and maintained by Hongbo Liu. For any question about SMART, you are welcome to contanct us (hongbo919@gmail.com).
If you use this tool, please cite the paper
Hongbo Liu et al. Systematic identification and annotation of human methylation marks based on bisulfite sequencing methylomes reveals distinct roles of cell type-specific hypomethylation in the regulation of cell identity genes Nucleic Acids Res: 2016 ,44(1) ,75-94.
1. SMART is developed based on QDMR which is a Shannon entropy-based software for identification of DMR from pre-defined regions of interest. QDMR provides visual interface which is useful for users without any programming basis.
2. SMART have been used to de novo identification of methylation marks of 42 humna cell types which is available at HumanMethlMark.

